Chapter 7 Your first PLINK tutorial
Learning outcomes: At the end of this chapter, you will be able to change genotype data formats with PLINK.
In the previous posts, you read about the general suggestions for the work environment, downloaded the PLINK software, and genotype data for a surprisingly large number of animals. But the program was not executed yet in any meaningful way... But now everything will change and you will finally run the PLINK program. Your goal will be to transform the binary file format saved as bim, bed, and fam files to a text-based genotype format saved as ped and map files. This exercise will also give you a detailed description of the use of PLINK. You will see that it is not difficult at all. Frankly, I look at the program as some kind of building-block game. You need to know what you want to achieve and all you need to do is add the correct elements to it. The first thing you need to write down is some sort of base structure. In fact, you can start with this very same line all the time and add elements to it.
But before you begin...
...I know, I know I am annoying with these recommendations all the time, and you are eager to jump in... But hear me out... You can type the PLINK commands directly to the command line, but don't do that. You will see that there will be many mistakes and re-runs all the time, and this way you will need to re-type all the time. This is a huge loss of time. You don't want that. Open a new text file instead and write your program script there. Ideally, this text file is saved in a cloud storage directory, so it is being automatically backupped upon save. Remember: the script files are very small in size, but extremely valuable given the amount of time you invested in writing them.
...one more piece of advice, in case you are new to scripting and programming. You might be surprised, but the scripts you write should be readable by the computer, but perhaps even more importantly by people, including future you. Let me explain... You write any script today and you sort of know what it does. I guarantee you that if you come back to it even after a week, you will have to spend quite some time figuring out what it does. Not to mention if you wrote some stuff like two years ago... Or imagine that you have to send this script to your colleague, who was not involved in the writing at all! If it is not clear how to change even basic things like input file names or locations, you are just looking for problems. So just document your code using plain words what some crucial lines or sections do. I use the # hashtag sign at the beginning of each comment line to indicate it as such. Also, lines starting with a # are ignored in many programs, so do not cause general errors.
Long story short: Document your code!
So now you will run PLINK. For real this time... Open the command prompt in a folder where you have the plink executable file and the genotype data, as described before in the PLINK - Software for genomic analyses chapter. Open a new text file and copy the following lines in there:
# Change binary genotype to ped+map format
plink --bfile ADAPTmap_genotypeTOP_20160222_full --cow --nonfounders --allow-no-sex --recode --out ADAPTmap_TOPSave the text file. From now on any change you implement will be written to the text file first, so you can adapt easily in case of need. Copy the whole plink line to the command prompt (without the comment line) and press enter. You have to have 1Gb free space for the recoded file. If everything went well, you will see this:
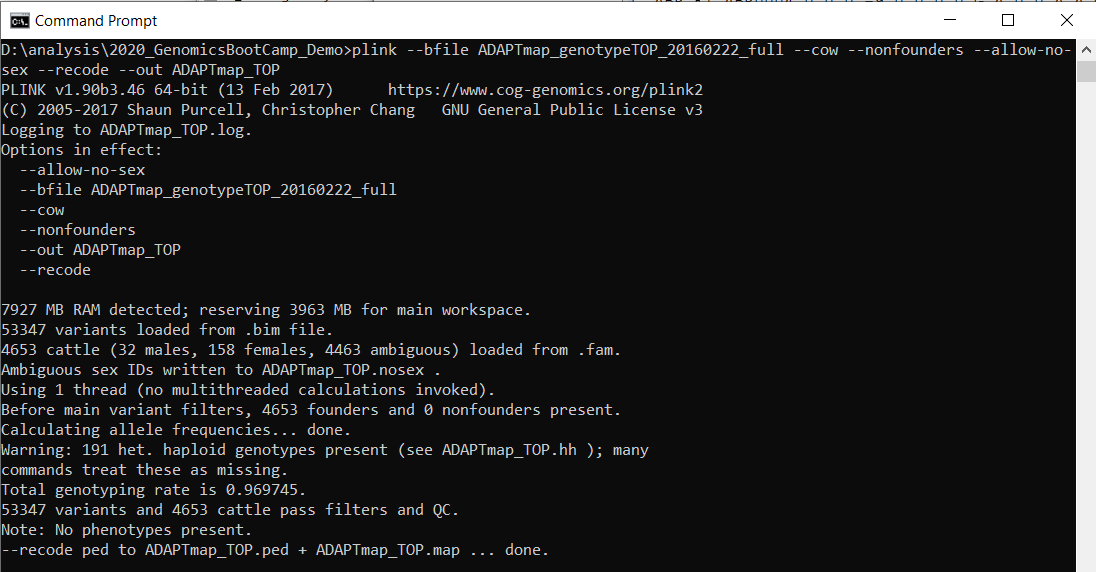
Figure 7.1: A successful PLINK run
In the following section I will explain what you just did in two parts:
First, let's start with the PLINK options. I will list them, simultaneously providing a link to them on the PLINK website. I will also tell you how can you (easily?) find answers for any PLINK option.
Second, I will talk about the resulting ped and map files, including their structure.
7.1 The PLINK options
I start with a general comment about the overall structure of PLINK that you have already noticed. After the program name, there are various options preceded by a double dash "--". Some less-appropriate-text-editors like MS Word might autocorrect this to a long dash, which will result in an error. So again, just use a proper text editor.
The PLINK options come in two formats:
--optinName1
--optionName2 space additionalParameter(s)RelatedToOptinName2
What the options used in the previous run mean:
--bfile ADAPTmap_genotypeTOP_20160222_full This is how you specify that your input file format is a binary ped file. Because you have all three files in the same directory as the PLINK executable, you only need to specify the file prefix, and the program will automatically utilize everything from the bim, bed, and fam files.
--cow This specifies the number of chromosomes in your data set. In case you do not tell anything about chromosome sets, the program will use the human genome as a default setting. Now you might have noticed that we deal with goats, but we specify --cow here. This time we played on the fact that bot cattle and goats have 60 chromosomes. In case you use another organism, you either specify that option, or a new setting using --chr-set
--nonfounders Do you remember when I talked about parental information in the fam file? This option is related to bypass any animals with missing parent information being treated as founders. Now, when you read the description on the website, you will also see that it is considered only in a few cases anyway. The thing is, however, that I tend to forget to use the handle in specific cases, and then spend a lot of time figuring out what went wrong. This is the pesky type of error when there is no error message, but the outcome is just incorrect. So to avoid all of this, I use --nonfounders in all my plink lines.
--allow-no-sex Similar to parental info, the info on sex is also many times missing. As it might or might not be problematic for certain analyses, I have decided to include this option in all my PLINK lines. The justification is the same as for --nonfounders.
--recode This very simple command is the one that actually does all the work. By putting this in your code you specify that you want to have ped and map files as output.
--out ADAPTmap_TOP Specifies the file name for newly created files.
So now you might have a very valid question: "This is all nice, but how do I find descriptions for other types of analyses I want to do?"
The two possibilities are:
- If you know exactly what are you looking for, you can use the handy search tool at the bottom of the left panel on the website, as shown in the picture. The result(s) below the search box are clickable links to the resource.
Figure 7.2: Find this bottom left on the PLINK website to get help
- If you don't know what you are looking for, try to explore the topics discussed on the left panel on the PLINK website, check other people's code, or simply ask colleagues how to do something. Alternatively, you can also try to fish for answers in the list of all PLINK options, using Ctrl+F text search capacities of your browser. You can also get there by clicking the "index" keyword link above the search bar.
Here I would note that PLINK can do many things, but certainly not everything. So do not expect to be a one-stop-shop for all of your genomic analysis needs. It is just a tool. A handy one, but still just one out of many.
7.2 The ped and map file format
In this section, we will take a brief look at the newly created files and tell something about their structure.
You might have noticed that there are a few new files created in the same directory you have run the program. From these files, the ones with file extension .ped and .map are the most important.
The .map file is very similar to the previously described .bim file, just without the last two columns with genotypes.
The .ped file structure is essentially the concatenation of the .fam file (one line per individual), followed by human-readable genotypes in text format. Every two columns represent one SNP in a space-delimited format.
To open the .map file should be no problem. The .ped file however is nearly 1Gb in size! I got the "File too big to be opened" error message with Notepad++, but the TextPad opened it without problems (after a bit of waiting time, might be machine-dependent, you will see a small progress bar bottom left).
7.3 How to run PLINK from R
As a practical demonstration of work with genomic data in R Studio, we will use PLINK example we discussed before in this chapter. With this, you will see the elements that need to be included to integrate the PLINK script to R and also prepare you for the grand finale of the first section - the PCA analysis.
The script we will be running is the following:
# clear workspace
rm(list = ls())
# set working directory
setwd("d:/analysis/2020_GenomicsBootCamp_Demo/")
# run PLINK QC
system("plink --bfile ADAPTmap_genotypeTOP_20160222_full --cow --nonfounders --allow-no-sex --recode --out ADAPTmap_TOP")From my personal experience in learning and teaching genomic data analysis to people of wide-ranging levels of experience, just telling you to copy-paste-and-run-the-script does not lead anywhere. So I will explain the most important elements of this script.
All lines starting with a hashtag # are comment lines. As I explained in a previous post, these are essential in all scripts. You need to be very clear what each part of the script is doing, which is much easier if you comment on it. The line rm(list = ls()) is a very useful one that I use in all my R scripts. It deletes everything from the workspace. This way I know that there are no pesky leftovers from previous analyses that might compromise my runs. (Ever had an experience of "The script was running before and it is not running now!"? This might be because of unintended data sets in the work environment.)
The setwd() is an R function that (as the name implies) sets the working directory for this session. The working directory is the place, where R will look for all the data for the analyses and place any output files if you do not specify otherwise. By default, this is a directory somewhere on your system drive. While it will work, I strongly suggest changing it to comply with your own file organization structure in a custom directory, as discussed before. You will need to change this for your run. Just put the PATH to your working directory containing PLINK and the genotype files between the quotation marks. Please note that R uses the opposite slashes as Windows.
You probably recognize the contents of the system() function. This is the exact copy of the PLINK command we used before, again between quotation marks. That's right! It is this easy to run PLINK from R. With the use of this function, the R opens the system command line, runs the line of code, and closes the command line.
Each line of the script above could be run separately. To do this, just put your cursor to the line you want to execute, and press the "Run" button in the top right corner. Alternatively, you can use the even better and arguably quicker method of simultaneously pressing Ctrl+Enter (Command+Enter in Mac).
7.4 Exercise
Phewww... You made it to the end of this unexpectedly long description! Congratulations! But to really drive home the message and lock the knowledge in your memory, I have a small task for you.
You see, the PLINK file formats are really popular, but there are many others out there. The good news is, that you can use PLINK to transform files to other popular formats. One of them is undoubtedly the so-called variant call format that is the standard output file from whole-genome sequencing pipelines, and a possible input to some other programs. So your task is to change the ADAPTmap file to vcf file format.
Hint: if I were you, I would explore the various options of the --recode option on the website. wink-wink
As always, you can compare your solution to the one on YouTube. The video also contains some bonus information on related problems you might face during analyses, so make sure to check it out regardless.
If the embedded video does not start, click it again to "Watch on YouTube". Direct link: https://www.youtube.com/watch?v=c1LSFiv9CxY